UMAPは高次元データを似たもの同士が近くなるように次元縮約してくれる便利な手法だ。 t-SNEよりも高速なことに加え、訓練しておいたモデルを新規データに適用できることも魅力。
そんなUMAPを異常検知の前処理に使うと、UMAPを使わない場合よりもうまく異常検知できるそうだ。例えば以下のリンクはPythonでMNISTデータセットに対してUMAP+LOFした例だ。
One-class SVMやLOFといった異常検知モデルでは正常データだけを学習し似ていないものは異常と判定する。その文脈からいくと、ついUMAPも正常データだけで学習させたくなってしまう。しかし、異常データのいくらかを学習に使えるなら使った方が、うまい縮約ができるのではないか、というのが今回の話題だ。
irisデータセットにおいて、setosa、versicolor、virginicaのいずれかを正常データとした時に、
- 正常データのみをUMAPで学習した場合
- 正常データに加え、もう一種をUMAPで学習した場合
- たとえばsetosaが正常データな場合はsetosa + versicolor、もしくはsetosa + virginicaで学習する
の各場合について、2次元に縮約した結果を可視化してみよう。実際には1000次元とかで次元の呪いの影響を受けて異常検知アルゴリズムがまともに機能しない場合を想定している。
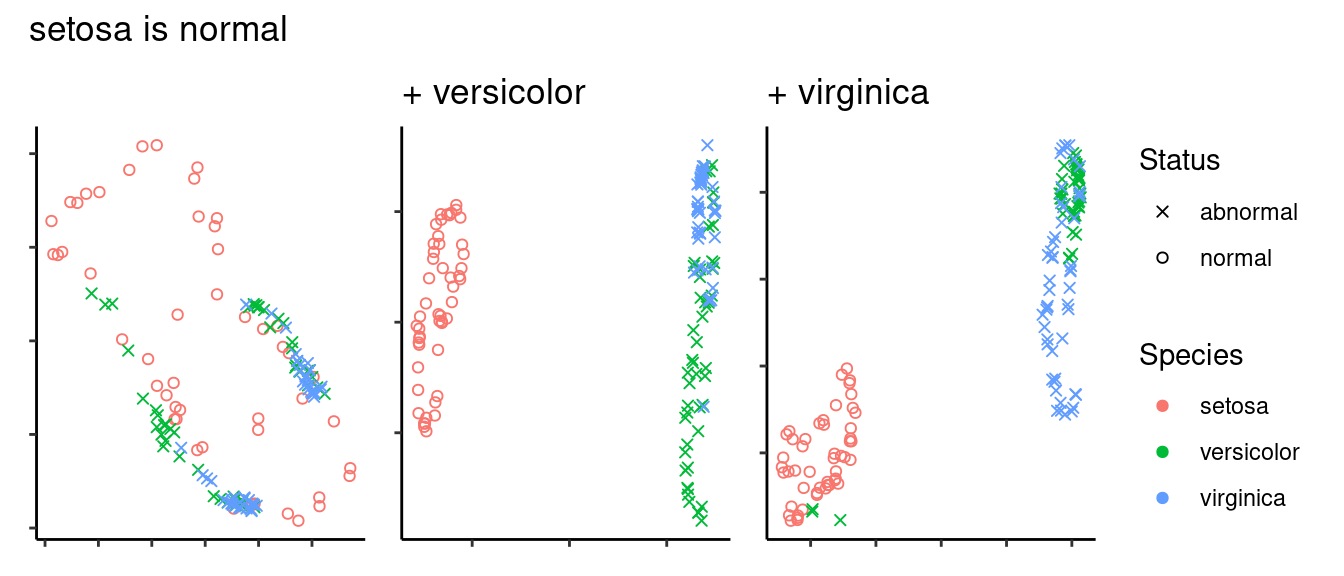
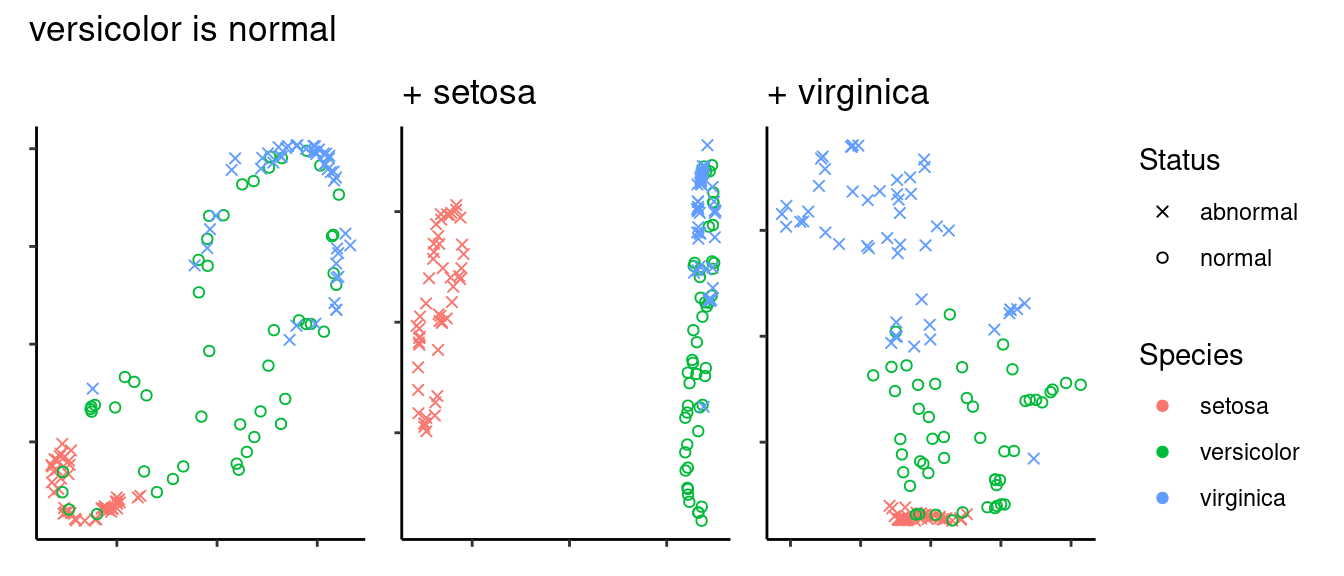
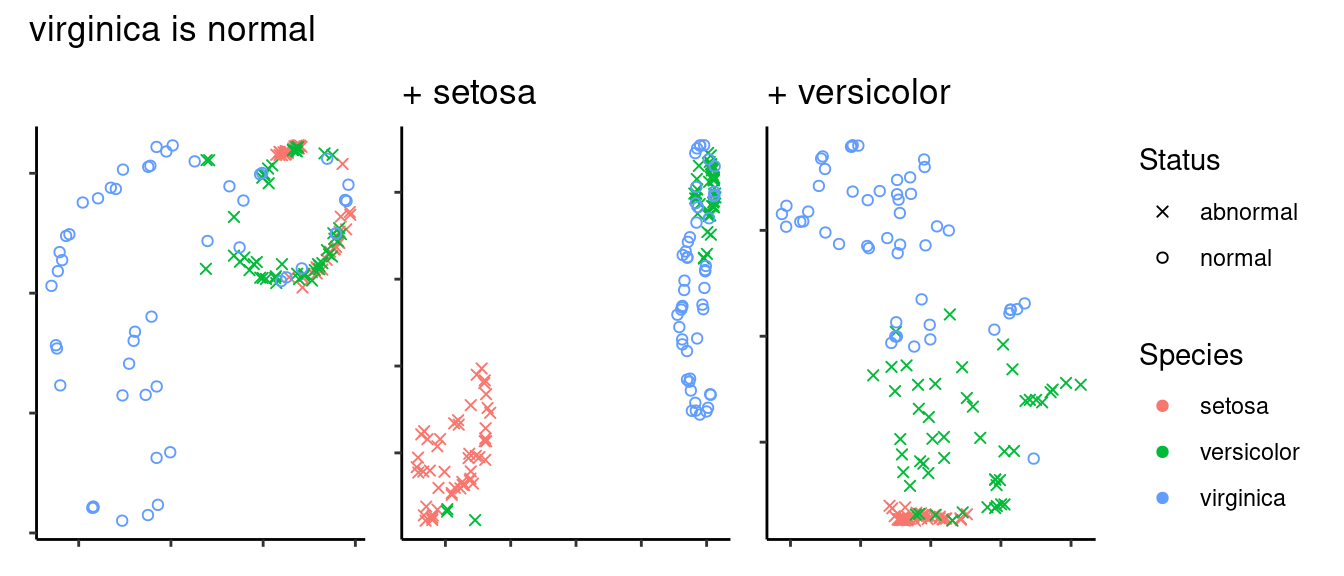
すると、いずれの種を正常データとした場合でも、いくらかNGデータも学習させてやった方がよさそうな見た目をしている。そこら辺は定量的に語れよって話ですが、力尽きたのでここまで。
- 正常データだけで学習すると、正常データ内の相違を要約してしまうため、正常データが分散してしまう
- なんだか数珠繋ぎになった正常データの一部に異常データが重なっている感じ
- パラメータ調整して局所?大局?構造を保つようにすれば良いかもしれない
- 異常データも学習すると、分解能が改善する
- setosaで顕著。元の次元でもsetosaはSepal.LengthとSepal.Widthの2次元プロットをするだけで他と識別できるので宜なるかな。
ちなみにversicolorとvirginicaは全データから2次元に縮約した場合でも識別は難しい。
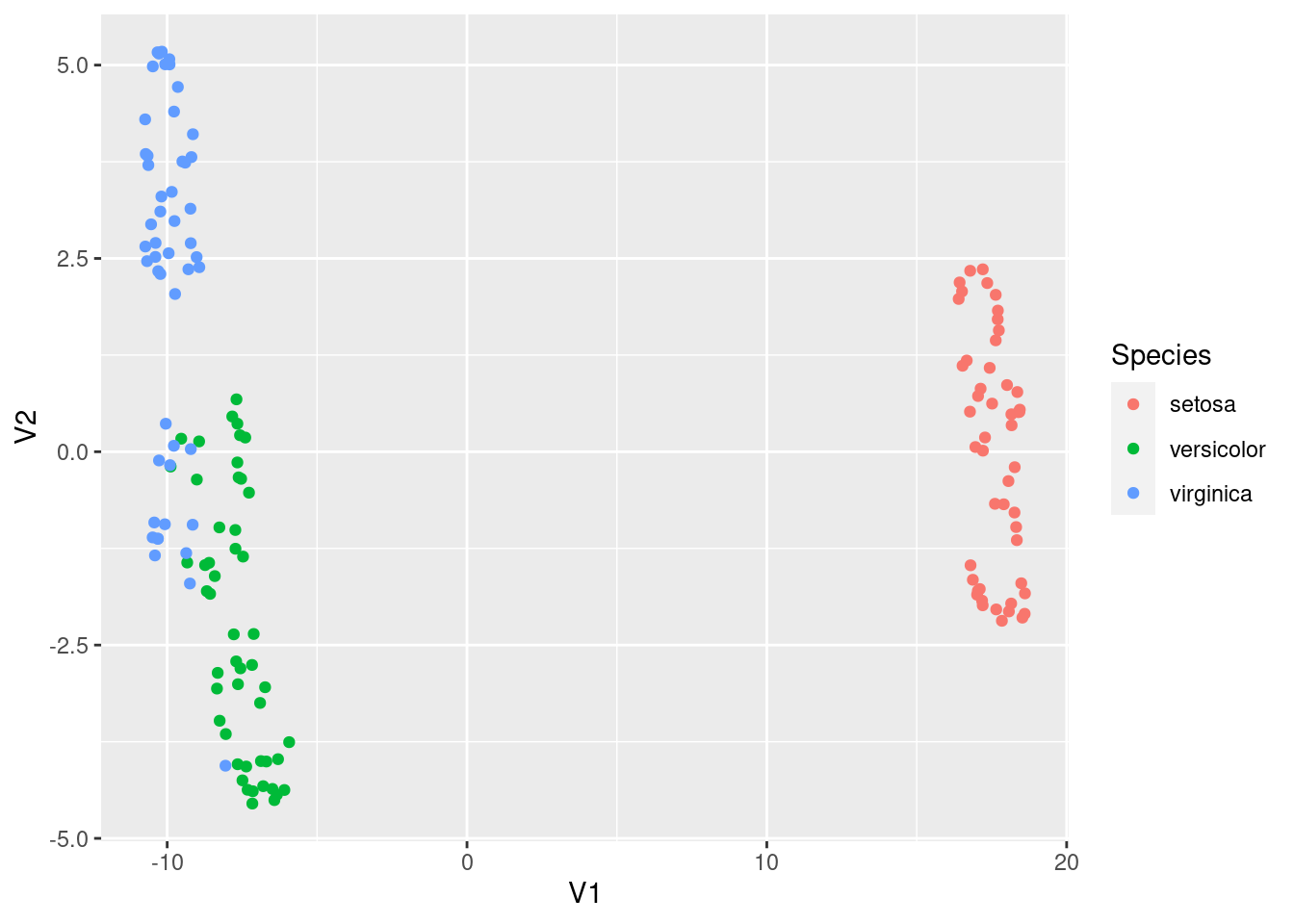
ソース
library(dplyr)
library(ggplot2)
library(purrr)
library(umap)
library(stringr)
set.seed(1)
# 選択した種においてUMAPを実施し
# すべての種を含めて可視化
plot_umap <- function(train) {
umap_fit <- umap::umap(iris[iris$Species %in% train, -5])
predict(umap_fit, iris[-5]) %>%
as.data.frame() %>%
mutate(
Species = iris$Species,
Status = ifelse(Species == train[1], 'normal', 'abnormal')
) %>%
ggplot() +
aes(V1, V2, color = Species, shape = Status) +
geom_point() +
scale_shape_manual(values = c(normal = "circle open", abnormal = "cross")) +
ggtitle(
if (length(train) == 1) {
NULL
} else {
paste('+', train[2])
}
) +
theme_classic() +
theme(axis.title = element_blank(), axis.text = element_blank())
}
# 正常データ扱いする種を指定し
# 単独でumapを学習した場合と、
# 異常値扱いの種を一つ加えて学習した場合を
# 比較
compare_train <- function(normal) {
species <- levels(iris$Species) %>% str_subset(normal, negate = TRUE)
patchwork::wrap_plots(
plot_umap(normal),
plot_umap(c(normal, species[1])),
plot_umap(c(normal, species[2]))
) +
patchwork::plot_annotation(title = paste(normal, "is normal")) +
patchwork::plot_layout(guides = "collect")
}
# compare_trainをirisの種ごとに実行
iris$Species %>%
levels %>%
map(compare_train) %>%
walk(print)set.seed(1)
umap::umap(iris[-5]) %>%
predict(., .$data) %>%
as.data.frame() %>%
mutate(Species = iris$Species) %>%
ggplot() +
aes(V1, V2, color = Species) +
geom_point()